




Jerome Kluza
- UFR DES SCIENCES DE SANTE ET DU SPORT
- FACULTE DE MEDECINE
Présentation
Dernières actualités
Le laboratoire recherche un étudiant de niveau Master M1 de formation Biologie cellulaire ou Biochimie pour réaliser un stage (minimum 4 semaines) au cours de l'année 2019/2020 sur nos thématiques de recherche.

Stratégies précliniques ciblant le métabolisme énergétique, les fonctions apoptotiques et la régulation du stress oxydant des mitochondries de cellules cancéreuses.
Nos recherches portent sur le rôle du métabolisme bioénergétique des cellules tumorales dans la résistance aux médicaments anticancéreux.
Les objectifs sont :
De caractériser le métabolisme mitochondrial et glycolytique des tumeurs solides (ex: mélanome) ou des hémopathies malignes (LAM, LMC) afin de déterminer de nouvelles cibles thérapeutiques.
De caractériser les modifications métaboliques au niveau cellulaire induites après traitements par des thérapies ciblées comme les inhibiteurs de la Serine/Thréonine kinase BRAF ou les inhibiteurs de la tyrosine kinase (Bcr-Abl ou de FLT3), puis d'identifier les modifications métaboliques acquises au cours de la résistance à ces traitements afin de développer des approches thérapeutiques basées sur ces altérations pour empêcher l'émergence ou éradiquer les cellules cancéreuses devenues résistantes.
Ces travaux m’ont amené à développer de nombreuses approches de biochimies métaboliques dans le cadre de mes recherches : Mesure de la phosphorylation oxydative mitochondriale (Seahorse, electrode de Clark), de la glycolyse, analyse métabolomique par RMN ou spectrométrie de masse, enzymologie (chaine respiratoire), dosage ATP, NADH, NADPH, microscopie électronique ou confocale des mitochondries.
Je dispense plusieurs enseignements sur le métabolisme des tumeurs: A l'Université de Lille: en M1 Biologie Santé (FST, Parcours Biologie Cellulaire, MC3 Biologie des cellules cancéreuses), en M1 Biologie Santé (Faculté de Médecine, BBG-05 Oncogenèse et Thérapies), en M2 Biologie Santé (JT14, physiopathologie et mitochondries); ainsi qu'à l'Université de Grenoble (Master de Biologie, Ingénierie de la santé M1: UE how to become a cancer cell).
J. Kluza

Mitochondrial metabolism, apoptotic function and oxidative stress in cancer cells: Towards Mitochondria-Targeted Preclinical Strategies
My research focuses on the role of bioenergetic metabolism of tumor cells in resistance to anticancer drugs.
The main objectives are:
Characterization of the mitochondrial and glycolytic metabolism of solid tumors (eg melanoma) or hematologic disease (AML, CML) to discover new therapeutic targets.
Characterization of cellular metabolic modification induced by targeted therapies such as serine/threonine kinase inhibitors (BRAF) or tyrosine kinase inhibitors (Bcr-Abl or FLT3), then identify the metabolic changes acquired in resistant cancer cells to develop therapeutic strategies.
These studies are conducted using mostly biochemistry approaches to assess : Mitochondrial oxidative phosphorylation (Seahorse, Clark's electrode), glycolysis, metabolomic analysis by NMR or mass spectrometry, enzymology of respiratory chain, ATP assay, NADH, NADPH, mitochondrial morphology (fluorescence or electron microscopy).
At Lille University, I provide several courses on tumor metabolism: in M1 Biology and Health science (FST, Cell Biology Pathology, MC3 Cancer Cell Biology), in M1 Biology and Health science (Faculty of Medicine , BBG-05 Oncogenesis and Therapies), in M2 Biology and Health science (JT14, physiopathology and mitochondria); as well as at the University of Grenoble (Master of Biology, Health Engineering M1: EU how to become a cancer cell).
La phosphorylation oxydative mitochondriale des blastes : biomarqueur prédicif de la survie des patients atteints de leucémies aigues myéloïdes

thèse financée par la Région des Hauts de France et l'Université de Lille
Quentin Fovez, thèse soutenue le 17/12/2021
quentin.fovez@inserm.fr
Les leucémies aiguës myéloïdes (LAM) constituent un ensemble d'hémopathies malignes caractérisées par l'expansion clonale dans la moelle osseuse de précurseurs de cellules sanguines bloqués à un stade précoce de leur différenciation, les blastes. Le diagnostic et le suivi de la maladie reposent sur différents examens biologiques et biochimiques des blastes du sang et de la moelle osseuse. La faible survie observée dans les LAM, 24% 5 ans après le diagnostic, s’explique entre-autre par le fait que même si deux tiers des patients répondent initialement au traitement, la majorité d’entre eux finiront par rechuter. L’orientation du choix thérapeutique repose sur l’évaluation de critères cytogénétiques et moléculaires qui conduisent à la classification en groupes pronostiques identifiés selon les recommandations de l’ELN. Cependant cette classification présente des limites. Pour améliorer sa prédictivité, de nouveaux biomarqueurs complémentaires doivent être identifiés et caractérisés. Au cours de cette thèse, nous avons dans un premier temps développé une méthodologie robuste et fiable pour mesurer la consommation d'oxygène de cellules leucémiques de patients atteints de LAM afin d'évaluer les différents états de la phosphorylation oxydative mitochondriale comme potentiel biomarqueur fonctionnel des LAM. Puis nous avons appliqué cette méthodologie pour mesurer les différents paramètres métaboliques des blastes issues du sang de 58 patients atteints de LAM isolées au moment du diagnostic.
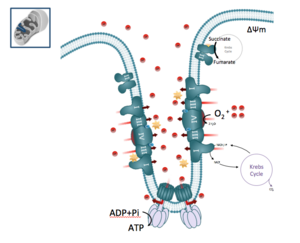
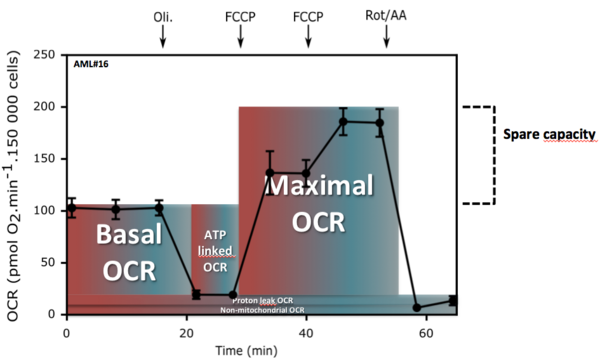
Nous avons d'abord développé un protocole pour mesurer, à l'aide du Seahorse XFe24, les différents états de la consommation d’oxygène mitochondriale de blastes isolés de la moelle ou du sang. Nous avons montré qu’il était possible de mesurer le métabolisme énergétique de ces cellules directement après le prélèvement, après 18 heures de cultures ou après la mise en culture suite à leur cryoconservation. Ces protocoles ont été développés afin de mesurer : la consommation d'oxygène mitochondriale (couplée ou non à la synthèse d'ATP) et la capacité de réserve respiratoire (SRC) qui correspond à la capacité des cellules à accélérer la chaine respiratoire après exposition au protonophore FCCP.
A partir d'une étude de cohorte prospective (58 patients), nous avons évalué les différents états de la consommation d'oxygène mitochondriale de blastes issues de sang de patients atteints de LAM. Les analyses biostatistiques, basées sur le calcul du risque relatif instantané (hazard ratio, HR), ont déterminé que SRC était significativement corrélée à la survie des patients. Après discrétisation de la SRC, nous avons établi un seuil distinguant deux groupes de patients. Les patients présentant des blastes avec une SRC supérieure à 75 pmol d’O2.min-1présentent une survie significativement plus élevée que les patients avec des blastes dont la SRC est inférieure à ce seuil.
Nous avons caractérisé les événements biochimiques associés à l'accélération de la chaîne respiratoire à partir de différentes lignées de LAM ou de blastes issus de patients. En utilisant des inhibiteurs pharmacologiques (BPTES, UK5099 et Etomoxir), des approches d'ARN interférence (knock-down MPC2) et des analyses de métabolomiques (après incubation avec de l’[U13C]-Glucose ou de l’[U13C]-Glutamine), nous avons démontré que l'oxydation du pyruvate via la pyruvate déshydrogénase était une voie majeure activée durant l’accélération de l’activité de la chaîne respiratoire induite par le FCCP.
En conclusion, nous avons démontré qu’il existait une hétérogénéité du métabolisme oxydati
f des blastes issues du sang de patients atteints de LAM et que la SRC permettait de distinguer deux groupes de patients dont la survie est significativement différente. Ces résultats établissent une base méthodologique solide utilisable en clinique et montrent l’intérêt de la SRC en tant que potentiel biomarqueur pour compléter les biomarqueurs actuels utilisés pour établir la classification ELN.

Thèse financée par l'Université de Lille
Claire Degand (D1 - 2021/2024)
claire.degand@inserm.fr
Résumé:
La persistance des cellules leucémiques est une limite majeure de l’efficacité des traitements à l’origine des récidives chez les patients atteints de leucémie aigue myéloïde (LAM). Après exposition aux traitements conventionnels, la survie des cellules leucémiques persistantes est soutenue principalement par le métabolisme énergétique mitochondrial. Ainsi plusieurs études précliniques ont montré que l’association d’inhibiteurs de la phosphorylation oxydative mitochondriale (tigécycline, metformine) avec divers traitements anticancéreux constitue in vitro une combinaison thérapeutique efficace pour éradiquer les cellules leucémiques survivantes. Evaluer l’activité des mitochondries de LAM issues de blastes de patients pourrait donc donner une information prédictive de la réponse aux traitements. Cependant la chaîne respiratoire des mitochondries fonctionne rarement au maximum de ses capacités et elle possède une réserve lui permettant d’augmenter son fonctionnement, notamment durant les épisodes de stress énergétiques : c’est la capacité de réserve mitochondrial. Cette composante métabolique peut être évaluée par différentes techniques d’oxymétrie à partir de prélèvements de blastes issus de sang ou de moelle de patients atteints de leucémie. Dans le cadre de ce projet, je propose de déterminer si la capacité de réserve mitochondriale est un potentiel biomarqueur de la réponse aux traitements des patients atteints de LAM. Puis je caractériserai les processus biochimiques et les acteurs impliqués dans le contrôle de cette composante métabolique. Enfin je déterminerai l’influence de la capacité de réserve sur la prolifération, l’agressivité des LAM, sur la survie dans des conditions de stress métaboliques et/ou sur la résistance aux médicaments anticancéreux.